
17/10/2024
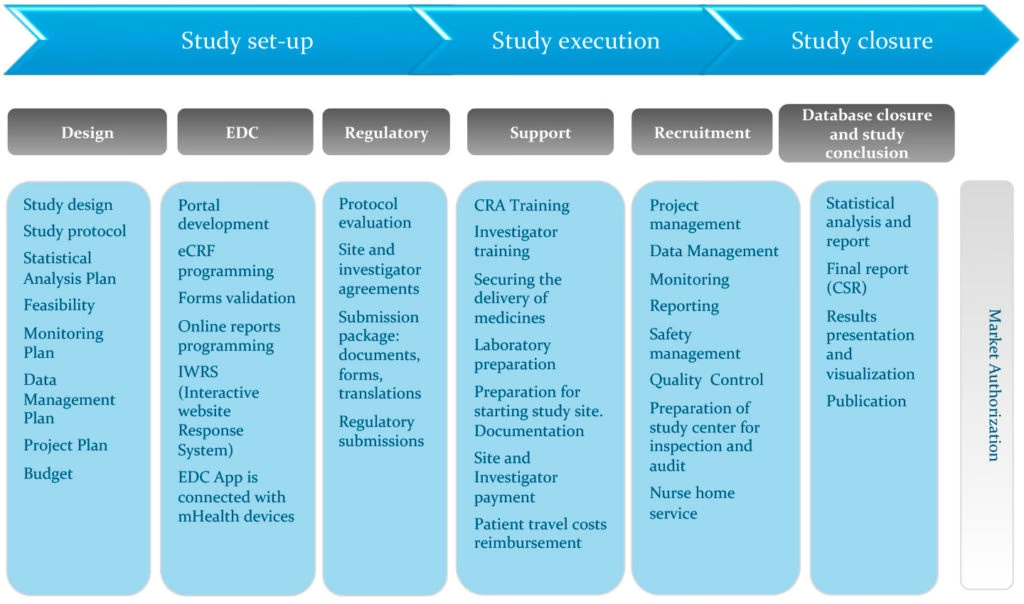
W dynamicznie rozwijającym się świecie badań klinicznych, audyty stanowią fundament zapewnienia jakości, bezpieczeństwa pacjentów i wiarygodności danych. W tym artykule zagłębimy się w istotę audytów badań klinicznych, prześledzimy ich ewolucję, omówimy nowoczesne podejścia oparte na ryzyku i zbadamy, jak stać się audytorem w tej kluczowej dziedzinie. Zrozumienie audytów badań klinicznych jest niezbędne dla każdego, kto jest zaangażowany w proces opracowywania nowych terapii i leków, od sponsorów i badaczy, po personel ośrodków badawczych i pacjentów.
https://www.youtube.com/watch?v=ygUSI3ByYWt0eWtha2xpbmljem5h
- Czym jest audyt w badaniach klinicznych?
- Jak ewoluowały audyty badań klinicznych
- Jak jeszcze bardziej rozwijamy audyty badań klinicznych
- Klucze do sukcesu
- Lista kontrolna audytu ośrodka badacza klinicznego
- Znaczenie niezależnych audytorów
- Kilka często zadawanych pytań dotyczących audytu ośrodka badawczego
- P: Jak podejście oparte na monitorowaniu opartym na ryzyku (RBM) wpływa na proces audytu ośrodków badaczy klinicznych?
- P: Jakie są implikacje elektronicznych systemów gromadzenia danych (EDC) na audyty ośrodków?
- P: W jaki sposób ustalenia z audytów ośrodków badaczy klinicznych są zazwyczaj przekazywane sponsorowi i ośrodkowi?
- P: Czy ośrodek badacza klinicznego może nie zdać audytu i jakie są tego konsekwencje?
- P: Jaką rolę odgrywają audytorzy w ciągłym doskonaleniu procesów badań klinicznych?
- P: Jak zarządzane są globalne badania kliniczne z wieloma międzynarodowymi ośrodkami z perspektywy audytu?
- Jak zostać audytorem medycznym? / Audytor wewnętrzny ISO 13485
- Czym jest norma ISO 13485?
- Ile zarabia audytor badań klinicznych?
Czym jest audyt w badaniach klinicznych?
Audyt ośrodka badawczego w badaniach klinicznych to formalne i systematyczne badanie sposobu prowadzenia badań klinicznych w ośrodkach badawczych. Jest to narzędzie zapewnienia jakości, którego celem jest ochrona praw, bezpieczeństwa i dobrostanu uczestników badania oraz zagwarantowanie, że dane generowane w badaniach klinicznych są dokładne, wiarygodne i weryfikowalne. Audyty te są przeprowadzane w celu oceny zgodności z protokołem badania, wytycznymi Dobrej Praktyki Klinicznej (GCP) i obowiązującymi wymogami regulacyjnymi.

Główne powody przeprowadzania audytów to:
- Ochrona praw i dobra uczestników badań.
- Zapewnienie wiarygodności i rzetelności danych z badań klinicznych.
- Weryfikacja zgodności z zatwierdzonym protokołem i jego zmianami.
- Ocena kwalifikacji i zdolności badaczy oraz personelu ośrodka do prowadzenia badania.
- Upewnienie się, że ośrodek przestrzega wymogów regulacyjnych i standardów GCP.
- Identyfikacja obszarów potencjalnego ryzyka i niezgodności.
- Dostarczanie informacji zwrotnej i wskazówek dotyczących ciągłego doskonalenia.
Kiedy przeprowadzane są audyty?
Audyty mogą być przeprowadzane na różnych etapach badania klinicznego:
- Audyty przed rozpoczęciem badania: przed rozpoczęciem badania, aby upewnić się, że ośrodek jest zdolny do jego przeprowadzenia.
- Audyty rutynowe: przeprowadzane w trakcie badania, w celu monitorowania bieżącej zgodności i jakości danych.
- Audyty celowe: wywoływane konkretnymi zdarzeniami lub obawami, które pojawiają się w trakcie badania.
- Audyty zamykające: przeprowadzane po zakończeniu badania, w celu przeglądu całościowego przebiegu badania i gromadzenia danych.
Jak przeprowadzane są audyty?
Audyt zazwyczaj obejmuje:
- Przegląd dokumentacji podstawowej, takiej jak Broszura Badacza, protokół badania, formularze świadomej zgody i formularze raportów przypadków (CRF).
- Wywiady z głównym badaczem, koordynatorami badania i innym personelem ośrodka.
- Bezpośrednia obserwacja procesów i procedur badania.
- Weryfikacja dokładności danych poprzez porównanie dokumentów źródłowych (np. dokumentacji medycznej) z danymi raportowanymi do sponsora.
- Ocena zgłaszania i postępowania z działaniami niepożądanymi.
- Ocena przechowywania i postępowania z produktami badanymi.
Co dzieje się po audycie?
Po audycie audytor sporządza raport szczegółowo opisujący ustalenia i obserwacje. W przypadku zidentyfikowania problemów z niezgodnością lub obszarów wymagających poprawy, ośrodek jest zazwyczaj zobowiązany do odpowiedzi poprzez działania naprawcze i zapobiegawcze (CAPA). Mogą być przeprowadzane audyty kontrolne, aby upewnić się, że działania CAPA są skutecznie wdrażane.
Jak ewoluowały audyty badań klinicznych
W przeszłości audyty kliniczne były przeprowadzane cyklicznie i obejmowały wszystkie aspekty procesu od początku do końca. Zamiast rutynowych audytów całego procesu, organy regulacyjne, w tym FDA, zalecają obecnie podejście oparte na ryzyku, aby określić czas i zakres audytów. To przesunięcie nacisku kładzie na efektywne wykorzystanie zasobów poprzez kierowanie ich na obszary, które stwarzają największe ryzyko organizacyjne.
Planowanie audytu opiera się obecnie na ocenach ryzyka, które uwzględniają czynniki operacyjne, regulacyjne i zgodności, aby zidentyfikować krytyczne obszary do przeglądu. Audytorzy koncentrują się na elementach, które wpływają na bezpieczeństwo pacjentów, względy etyczne i integralność danych, kierując się analizą danych i wskaźnikami ryzyka. Ta metoda zwiększa efektywność organizacyjną poprzez koncentrację na obszarach o największym potencjalnym ryzyku.
Jak jeszcze bardziej rozwijamy audyty badań klinicznych
Naszym celem jest rozwój audytów opartych na ryzyku poprzez zastosowanie tego podejścia do identyfikacji i raportowania ustaleń audytowych. Ta metoda pozwala nam podkreślić niezgodności i wskazać wadliwe procesy, które powodują te problemy. Łącząc niedociągnięcia bezpośrednio z ich konsekwencjami, wzmacniamy argumentację za koniecznymi zmianami i ulepszeniami, ułatwiając zainteresowanym stronom jaśniejsze zrozumienie potrzeby działania.
Przykład:
Rozważmy przypadek, w którym firma farmaceutyczna konsekwentnie napotyka problemy z integralnością danych podczas badań klinicznych. Problemy te często plasują się wśród najważniejszych ustaleń podczas inspekcji regulacyjnych i audytów wewnętrznych. Typowy audyt może ujawnić, że dane z badań nie były rejestrowane na czas, co prowadzi do działań naprawczych skoncentrowanych na bieżących praktykach postępowania z danymi, takich jak przeszkolenie personelu w zakresie prawidłowych procedur dokumentacyjnych. Jednak bardziej wnikliwe podejście polegałoby na zbadaniu podstawowych procesów przyczyniających się do tych problemów z integralnością danych. Na przykład, można odkryć, że elektroniczny system gromadzenia danych (EDC) nie jest przyjazny dla użytkownika, co powoduje opóźnienia w wprowadzaniu danych. Alternatywnie, przyczyną źródłową może być niedostateczna obsada personelu lub brak jasnych protokołów zarządzania danymi. Poprzez zidentyfikowanie podstawowych wad procesów - czy to technologicznych, proceduralnych, czy związanych z zasobami - działania CAPA można zaprojektować w celu poprawy ogólnego systemu zarządzania jakością. Mogłoby to prowadzić do ulepszeń w systemie EDC, zmiany protokołów zarządzania danymi lub dostosowania modeli obsady personelu, zapobiegając w ten sposób podobnym problemom w wielu badaniach klinicznych i ośrodkach.
Ocena systemu zarządzania jakością (QMS) zarówno w ośrodku badacza, jak i na poziomie sponsora ma kluczowe znaczenie dla identyfikacji miejsc, w których mogły wystąpić błędy w całym cyklu życia badania klinicznego.
Oto, jak audytor może podejść do tej oceny:
- Na poziomie ośrodka badacza
- Etap planowania: Czy badanie zostało prawidłowo skonfigurowane? Jeśli na etapie planowania wystąpiły problemy, takie jak nieodpowiednie szkolenie lub niejasne protokoły, może to prowadzić do problemów systemowych w późniejszym czasie.
- Etap realizacji: Jak dobrze badanie zostało wykonane? Błędy na tym etapie mogą obejmować nieprawidłowe gromadzenie danych lub nieprzestrzeganie protokołu badania.
- Etap kontroli: Czy główny badacz posiadał odpowiedni nadzór i mechanizmy kontroli, aby wcześnie wychwycić problemy? Brak skutecznego monitorowania może spowodować, że błędy pozostaną niezauważone i nierozwiązane.
- Etap działania: Czy podjęto szybkie i skuteczne działania naprawcze w celu rozwiązania zidentyfikowanych błędów? Brak działań lub nieskuteczne działania mogą pogorszyć problemy i prowadzić do dalszej niezgodności.
- Na poziomie sponsora
- Etap planowania: Czy narzędzia i instrukcje dostarczone przez sponsora były odpowiednie do pomyślnego wykonania? Niewystarczające wytyczne mogą prowadzić do zamieszania i niezgodności w ośrodku badawczym.
- Etap realizacji: Czy monitorowanie ośrodka badania klinicznego przez sponsora było skuteczne? Niewystarczające monitorowanie może przeoczyć krytyczne problemy, które zagrażają integralności danych i bezpieczeństwu uczestników.
- Etap kontroli: Czy istniały wskaźniki wydajności umożliwiające identyfikację ryzyka? Brak takich wskaźników może uniemożliwić wczesne wykrycie potencjalnych problemów.
- Etap działania: Jak skuteczne były mechanizmy kontroli sponsora w zapobieganiu lub ograniczaniu ryzyka? Nieskuteczne mechanizmy kontroli mogą nie zapewniać odpowiedniej ochrony przed ponownym wystąpieniem problemów.
Klucze do sukcesu
Aby podejście oparte na ryzyku w zarządzaniu jakością w formułowaniu ustaleń audytowych było skuteczne, wymaganych jest kilka kluczowych elementów:
- Dojrzała kultura jakości: Sukces tego podejścia zależy od dojrzałej kultury jakości w organizacji sponsora i jej partnerów. Zainteresowane strony muszą być skłonne do przyjęcia kompleksowej perspektywy, która wykracza poza granice działów, wspierając otwartość na ciągłe doskonalenie.
- Globalni właściciele procesów (GPO): GPO mają kluczowe znaczenie w globalnym modelu operacyjnym. Nadzorują procesy w granicach funkcjonalnych i geograficznych, zapewniając spójność i ustalając jasne oczekiwania. Są na bieżąco z zewnętrznymi i wewnętrznymi zmianami, które mogą wpływać na procesy i ułatwiają niezbędne adaptacje.
- Przywództwo funkcjonalne: Liderzy zespołów funkcjonalnych muszą być gotowi do zajęcia się ustaleniami audytowymi, które wskazują na braki w wykonaniu. Odgrywają kluczową rolę we wdrażaniu działań naprawczych na poziomie operacyjnym.
- Szkolenie audytorów: Audytorzy powinni być przeszkoleni w zakresie rozróżniania między przeprowadzaniem audytów opartych na procesach a przeprowadzaniem analizy przyczyn źródłowych. Muszą zrozumieć, że ich rolą jest identyfikacja, gdzie i dlaczego proces zawiódł, a nie samodzielne przeprowadzanie analizy przyczyn źródłowych.
- Badanie i ocena: Personel ośrodka badawczego i personel sponsora są odpowiedzialni za badanie przyczyn źródłowych awarii, biorąc pod uwagę czynniki związane z ludźmi, procesami, sprzętem lub systemami.
- Solidne działania CAPA: Koncentracja na wadach procesów, a nie na obwinianiu jednostek, prowadzi do solidniejszych działań CAPA. Ustalenia, które jasno określają problem procesu i jego wpływ, ułatwiają akceptację i umożliwiają ukierunkowane działania naprawcze.
- Rozwiązania na poziomie procesów: Poprzez identyfikację, który proces zawiódł, odpowiedni GPO może kierować procesem CAPA, zapewniając, że rozwiązania są wdrażane na odpowiednim poziomie w celu skutecznej zmiany.
- Ciągłe doskonalenie: Podejście powinno promować ciągłe doskonalenie, a audytorzy powinni być uznawani za ekspertów w myśleniu procesowym. Sprzyja to merytorycznym dyskusjom z audytowanymi, koncentrując się na ryzyku, wpływie i krytycznym myśleniu.
Lista kontrolna audytu ośrodka badacza klinicznego
Aby zapewnić zgodność z FDA podczas badań klinicznych, firmy powinny posiadać kompleksową listę kontrolną audytu. Oto strukturalna lista kontrolna z kluczowymi obszarami do przeglądu. Należy pamiętać, że nie jest ona wyczerpująca.
| Obszar kontroli | Opis |
|---|---|
| Zgodność z protokołem | Sprawdź, czy badacz i personel ośrodka przestrzegali protokołu badania zatwierdzonego przez IRB. |
| Institutional Review Board (IRB) | Potwierdź dokumentację i przechowywanie na miejscu wszystkich zatwierdzeń IRB, poprawek i dokumentów świadomej zgody. |
| Dokumentacja uczestników badania | Przejrzyj formularze świadomej zgody, dokumentację medyczną i inne dokumenty źródłowe pod kątem kompletności i zgodności z protokołem. |
| Inna dokumentacja badania | Sprawdź akta administracyjne, korespondencję, listy uczestników, dzienniki i formularze pod kątem dokładności i kompletności. |
| Świadoma zgoda uczestników badania | Upewnij się, że świadoma zgoda została uzyskana przy użyciu formularzy zatwierdzonych przez IRB i że proces ten jest udokumentowany. |
| Ujawnienie informacji finansowych | Przejrzyj oświadczenia o interesach finansowych od badaczy i personelu badania pod kątem aktualności. |
| Dokumentacja elektroniczna i podpisy elektroniczne | Sprawdź, czy systemy elektroniczne spełniają wymogi regulacyjne, a personel jest przeszkolony w zakresie ich obsługi. |
| Kontrola badanego produktu | Potwierdź prawidłowy odbiór, etykietowanie, inwentaryzację, przechowywanie i utylizację leku. |
| Przechowywanie i archiwizacja dokumentacji | Upewnij się, że dokumentacja jest przechowywana i archiwizowana zgodnie z protokołem i przepisami. |
| Raporty dla sponsora | Zweryfikuj przesyłanie wszystkich wymaganych raportów, zwłaszcza dotyczących kwestii bezpieczeństwa lub odstępstw od protokołu, do sponsora. |
| Kwalifikacje i umowy badacza | Oceń doświadczenie, przeszkolenie i wiedzę badacza na temat GCP i wymogów regulacyjnych. |
| Odpowiednie zasoby | Potwierdź, że badacz ma wystarczającą liczbę wykwalifikowanego personelu i obiektów do prowadzenia badania. |
| Opieka medyczna nad uczestnikami badania | Upewnij się, że uczestnicy badania otrzymują właściwą opiekę medyczną i nadzór nad decyzjami związanymi z badaniem. |
| Komunikacja z IRB | Sprawdź udokumentowane zatwierdzenia IRB dla wniosku o badanie, formularzy zgody i informacji dla uczestników. |
| Procedury randomizacji i zaślepienia | Przejrzyj zgodność z procedurami randomizacji i zaślepienia oraz dokumentację dotyczącą ewentualnego odślepienia. |
| Dokumentacja i raporty | Upewnij się, że dane raportowane sponsorowi są dokładne, kompletne, czytelne i terminowe. |
| Raporty o postępach | Potwierdź przesyłanie statusu badania do IRB i natychmiastowe zgłaszanie sponsorowi poważnych zdarzeń niepożądanych. |
| Podstawowe dokumenty regulacyjne | Przejrzyj dokumentację regulacyjną pod kątem kompletności, w tym formularz FDA 1572, protokół i korespondencję z IRB. |
| Personel badania | Zweryfikuj kwalifikacje, licencje i szkolenie całego personelu badania. |
| Monitorowanie danych i bezpieczeństwa | W przypadku badań wysokiego ryzyka, przejrzyj raporty DSMB pod kątem problemów z zgodnością lub wzorców. |
| Archiwizacja dokumentacji | Sprawdź, czy wszystkie dokumenty i dane są zorganizowane i bezpiecznie przechowywane. |
| Przedwczesne zakończenie lub zawieszenie badania | Upewnij się, że istnieją procedury informowania uczestników, organów regulacyjnych i dotyczące dalszej opieki nad uczestnikami. |
| Raport końcowy badacza | Zweryfikuj, czy IRB i organy regulacyjne zostały poinformowane o wyniku badania i czy wszystkie wymagane raporty zostały przesłane. |
Znaczenie niezależnych audytorów
Przeprowadzenie niezależnego audytu jest niezbędne z kilku powodów. Obiektywna ocena jest najważniejsza z nich. Niezależne audyty zapewniają świeże, bezstronne spojrzenie na systemy jakości programu, zapewniając dokładną i bezstronną ocenę.
Inne powody to:
- Unikanie konfliktów interesów: Audyty przeprowadzane przez strony związane z programem mogą nieumyślnie przeoczyć lub zbagatelizować problemy z powodu nieodłącznych konfliktów interesów.
- Zgodność z przepisami: Niezależne audyty mają kluczowe znaczenie dla proaktywnego wykrywania problemów, umożliwiając wprowadzenie poprawek, zanim zostaną one zidentyfikowane przez organy regulacyjne, takie jak FDA, podczas ich inspekcji.
- Zgodność dostawców: Przy znaczących inwestycjach związanych z opracowywaniem leków, zapewnienie, że dostawcy przestrzegają standardów jakości, ma kluczowe znaczenie. Niezależne audyty analizują procesy dostawców, aby upewnić się, że spełniają oni wymagania dotyczące zgodności, chroniąc przed kosztownymi błędami, które mogłyby zostać wychwycone przez FDA.
- Minimalizacja ryzyka: Poprzez wczesne identyfikowanie i rozwiązywanie luk w systemach jakości, niezależne audyty pomagają minimalizować ryzyko niezgodności, które może prowadzić do poważnych konsekwencji, takich jak unieważnienie badania lub nakazane działania naprawcze.
- Ochrona inwestycji: Niezależne audyty są proaktywnym krokiem w ochronie programu rozwoju leków i inwestycji finansowych poprzez zapewnienie, że wszystkie aspekty programu są zgodne z przepisami i na dobrej drodze.
- Rozwój systemu jakości: Firmy takie jak nasza nie tylko przeprowadzają niezależne audyty, ale także pomagają w opracowywaniu solidnych systemów jakości i wskaźników do monitorowania bieżącej zgodności, co dodatkowo chroni inwestycję.
- Wiedza specjalistyczna i spójność: Niezależne audyty są przeprowadzane przez doświadczonych specjalistów, którzy wnoszą wiedzę specjalistyczną i spójność do procesu audytu, przyczyniając się do wiarygodności i integralności działań na rzecz zgodności.
Aby zapewnić kompleksową zgodność i chronić program rozwoju leków, zawsze zalecamy korzystanie z usług wyspecjalizowanej firmy w zakresie niezależnego audytu.
Kilka często zadawanych pytań dotyczących audytu ośrodka badawczego
P: Jak podejście oparte na monitorowaniu opartym na ryzyku (RBM) wpływa na proces audytu ośrodków badaczy klinicznych?
RBM dostosowuje proces audytu do priorytetowego traktowania najbardziej krytycznych i wysokiego ryzyka aspektów badań klinicznych. W podejściu RBM audytorzy dają pierwszeństwo danym i procesom, które mają kluczowe znaczenie dla ochrony uczestników badań i zapewnienia wiarygodności wyników badań. Chociaż tradycyjne kompleksowe audyty mogą nadal występować, RBM prowadzi do bardziej efektywnej alokacji zasobów audytowych, koncentrując się na obszarach takich jak świadoma zgoda, przestrzeganie protokołu i integralność raportowanych danych. Audytorzy wykorzystują kombinację technik monitorowania na miejscu i scentralizowanego, aby identyfikować i minimalizować ryzyko w trakcie badania.
P: Jakie są implikacje elektronicznych systemów gromadzenia danych (EDC) na audyty ośrodków?
Systemy EDC znacząco zmieniły krajobraz zarządzania danymi klinicznymi, a co za tym idzie, proces audytu. Audytorzy muszą być teraz biegli w walidacji systemów elektronicznych, aby upewnić się, że są one zgodne z 21 CFR Part 11 i innymi odpowiednimi przepisami. Obejmuje to weryfikację bezpieczeństwa systemu, dokładności transferów danych i integralności dokumentacji elektronicznej. Audytorzy muszą również ocenić, w jaki sposób personel ośrodka jest przeszkolony w zakresie tych systemów oraz w jaki sposób zarządzane są poprawki danych i ścieżki audytu.
P: W jaki sposób ustalenia z audytów ośrodków badaczy klinicznych są zazwyczaj przekazywane sponsorowi i ośrodkowi?
Ustalenia audytu są zazwyczaj podsumowywane w formalnym raporcie z audytu, który jest udostępniany zarówno sponsorowi, jak i ośrodkowi. Raport będzie kategoryzował ustalenia zgodnie z ich dotkliwością, taką jak krytyczna, poważna lub drobna niezgodność. Będzie również zawierał zalecenia dotyczące działań CAPA. Ośrodek jest zazwyczaj zobowiązany do ustosunkowania się do ustaleń, określając, w jaki sposób rozwiąże każdy problem i zapobiegnie jego ponownemu wystąpieniu. Sponsor jest odpowiedzialny za zapewnienie, że ośrodek wdroży te działania CAPA i może przeprowadzić audyty kontrolne w celu weryfikacji zgodności.
P: Czy ośrodek badacza klinicznego może nie zdać audytu i jakie są tego konsekwencje?
Tak. Jeśli ośrodek nie zda audytu i zostaną stwierdzone istotne problemy z niezgodnością, które mogłyby wpłynąć na bezpieczeństwo uczestników, integralność danych lub naruszyć wymogi regulacyjne, może to mieć poważne konsekwencje. Konsekwencje mogą obejmować konieczność odpowiedzi za pomocą działań CAPA, aż po poważniejsze działania, takie jak zawieszenie badania w ośrodku, dyskwalifikacja badacza lub zgłoszenie ustaleń organom regulacyjnym, co może prowadzić do dalszych działań regulacyjnych.
P: Jaką rolę odgrywają audytorzy w ciągłym doskonaleniu procesów badań klinicznych?
Audytorzy odgrywają kluczową rolę w ciągłym doskonaleniu, nie tylko identyfikując niezgodności i obszary wymagające poprawy, ale także dzieląc się najlepszymi praktykami i przekazując informacje zwrotne na temat procesów. Ich ustalenia mogą pomóc w kształtowaniu programów szkoleniowych, udoskonalaniu protokołów i poprawie ogólnego przebiegu badań. Celem audytu jest nie tylko znalezienie błędów, ale pomoc ośrodkowi w ulepszeniu jego procesów na potrzeby przyszłych badań.
P: Jak zarządzane są globalne badania kliniczne z wieloma międzynarodowymi ośrodkami z perspektywy audytu?
Globalne badania kliniczne stwarzają wyjątkowe wyzwania ze względu na zróżnicowane wymogi regulacyjne i różnice kulturowe. Audytorzy muszą być zaznajomieni z lokalnymi przepisami każdego kraju uczestniczącego w badaniu. Audyty mogą być przeprowadzane przez lokalne zespoły z globalną koordynacją w celu zapewnienia spójności. Plan audytu dla takich badań jest często opracowywany z globalnym zakresem, ale dopuszcza lokalną elastyczność w celu uwzględnienia specyficznych wymagań i ryzyk regionalnych.
Jak zostać audytorem medycznym? / Audytor wewnętrzny ISO 13485
Szkolenie na Audytora Wewnętrznego ISO 13485 oferowane jest osobom z branży wyrobów medycznych, które przygotowują się do roli audytora wewnętrznego systemu zarządzania jakością, a także chcących zapoznać się z wymaganiami norm PN-EN ISO 13485:2016, PN-EN ISO 19011:2018. Aby zostać audytorem wewnętrznym systemu zarządzania jakością w organizacji, w której wdrożony jest system zarządzania wg ISO 13485, należy dobrze znać wymagania i zastosowanie tej normy. Audytor wewnętrzny może zostać powołany przez najwyższe kierownictwo spośród pracowników, ale powinien przejść specjalistyczne przeszkolenie z zakresu funkcjonowania systemu zarządzania jakością ISO 13485 i najlepiej, gdyby posiadał certyfikat ukończenia profesjonalnego kursu, np. Audytor wewnętrzny ISO 13485.
Czym jest norma ISO 13485?
ISO 13485 to norma, która określa wymagania systemu zarządzania jakością dla producentów, dystrybutorów, firm serwisujących i logistycznych w branży wyrobów medycznych i wyrobów medycznych do diagnostyki in vitro. Standard ten wywodzi się z międzynarodowych norm dotyczących zarządzania jakością i koncentruje się głównie na zachowaniu zgodności z przepisami. Podstawą koncepcji ISO 13485 jest model procesowy ISO 9001, czyli PDCA (Plan–Do–Check–Act). Norma PN-EN ISO 13485:2016 nakłada na kierownictwo organizacji obowiązek zapewnienia niezbędnych szkoleń w celu budowania świadomości zaangażowanego personelu, by usługi świadczone przez daną organizację spełniały założone standardy. Szkolenia takie jak Audytor Wewnętrzny ISO 13485, kończące się certyfikatem, są istotnym dowodem na spełnienie wymagań normy dotyczących szkoleń i ciągłego doskonalenia organizacji.
Ile zarabia audytor badań klinicznych?
Informacje o dokładnych zarobkach audytorów badań klinicznych nie zostały bezpośrednio zawarte w dostarczonym tekście. Zarobki audytorów badań klinicznych mogą być zróżnicowane i zależą od wielu czynników, takich jak doświadczenie, poziom wykształcenia, typ pracodawcy (firma farmaceutyczna, CRO, firma konsultingowa), lokalizacja geograficzna oraz zakres obowiązków. Audytorzy z większym doświadczeniem i specjalistyczną wiedzą, szczególnie w obszarze GCP i regulacji, mogą zazwyczaj liczyć na wyższe wynagrodzenie. Dodatkowo, posiadanie certyfikacji w zakresie audytu i znajomość norm takich jak ISO 13485 może również pozytywnie wpłynąć na potencjalne zarobki.
Podsumowując, audyty badań klinicznych są kluczowym elementem zapewnienia jakości i bezpieczeństwa w procesie opracowywania nowych leków i terapii. Od ewolucji podejścia do audytów, poprzez wdrażanie metod opartych na ryzyku, aż po znaczenie niezależnych audytorów i rolę norm ISO - zrozumienie tych aspektów jest niezbędne dla wszystkich uczestników branży badań klinicznych. Inwestycja w solidne systemy audytowe i wykwalifikowanych audytorów to inwestycja w przyszłość bezpiecznej i skutecznej medycyny.
Jeśli chcesz poznać inne artykuły podobne do Audyt badań klinicznych: klucz do jakości i bezpieczeństwa, możesz odwiedzić kategorię Audyt.